
Cyclic peptides are an increasingly important drug modality, valued for their potency, selectivity, and metabolic stability. By enforcing conformational control, cyclization enables effective engagement of complex targets such as protein–protein interactions. Advances in synthesis and design now allow discovery teams to engineer cyclic peptides with drug‑like properties, supporting a clear path from hit identification to clinical candidate. This whitepaper outlines the key design principles, synthetic strategies, and developability considerations that enable efficient discovery and optimization of cyclic peptide therapeutics.
Peptides occupy a distinct chemical space between small molecules and biologics. They offer interaction surfaces large enough to engage extended or shallow binding pockets while retaining synthetic accessibility and tunability.
From a discovery standpoint, this balance is critical since, small molecules often fail to disrupt protein–protein interactions due to insufficient contact area, while biologics may suffer from delivery limitations. Cyclic Peptides overcome these challenges by presenting pre-organized pharmacophores in a compact, chemically defined scaffold, improving both binding efficiency and selectivity.
The defining feature of cyclic peptides is conformational restriction imposed by macrocyclization. This structural constraint has several mechanistically important consequences:
These effects collectively translate into improved in vitro potency and increased functional durability in biological systems.
Cyclic peptides are widespread in nature and provide valuable insights into structure–function relationships. Because they have evolved to withstand harsh biological environments while maintaining high potency, they serve as ideal molecular frameworks for modern peptide engineering. As illustrated in Figure 1, these natural macrocycles participate in diverse biological functions—including defense, signaling, and enzyme inhibition—revealing how cyclic architectures can encode remarkable stability and specificity.
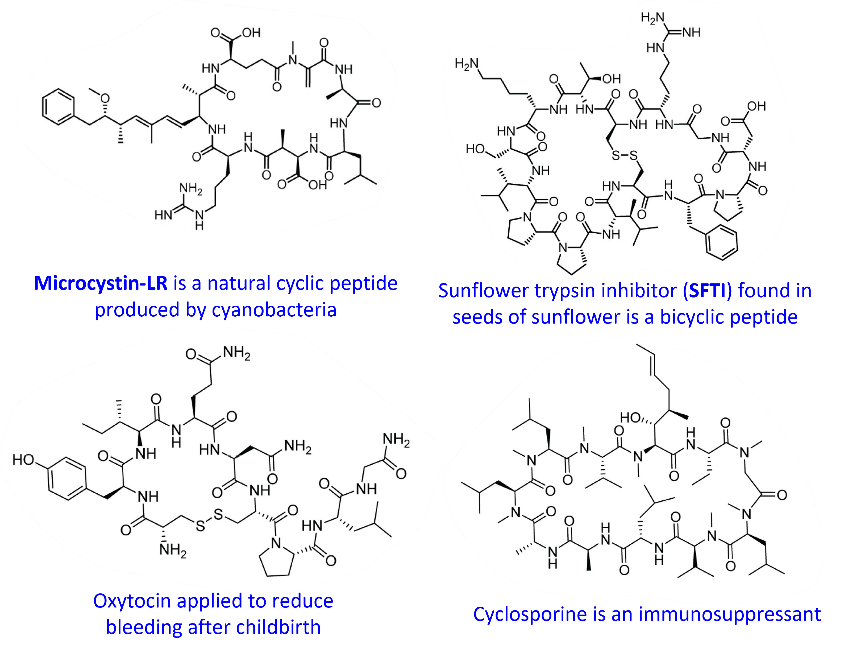
Figure 1: Natural cyclic peptides and their key biological functions.
Building on the concept introduced above—that natural cyclic peptides exemplify how macrocyclic structure dictates biological function—cyanobacterial toxins illustrate how even subtle architectural changes can dramatically shift biological outcomes. Conversely, beneficial macrocycles such as nisin highlight how constrained peptide scaffolds can achieve potent and selective antibacterial activity. Together, these examples provide discovery chemists with evolution‑validated templates for designing stable and biologically active macrocycles.
Cyclic peptides have delivered some of the most successful antibiotics used today. Glycopeptide antibiotics such as vancomycin and its next-generation derivatives exemplify how large, rigid macrocycles can recognize highly conserved bacterial cell wall motifs with exceptional affinity and specificity. Their complex architectures, produced through non-ribosomal biosynthesis, underscore the power of conformational control in overcoming resistance mechanisms (Figure 2).
In oncology, cyclic peptides such as RGD-based macrocycles exploit multivalent interactions with integrins and other tumor-associated receptors. Their constrained geometry allows precise spatial presentation of binding motifs, improving receptor discrimination and reducing off-target effects.
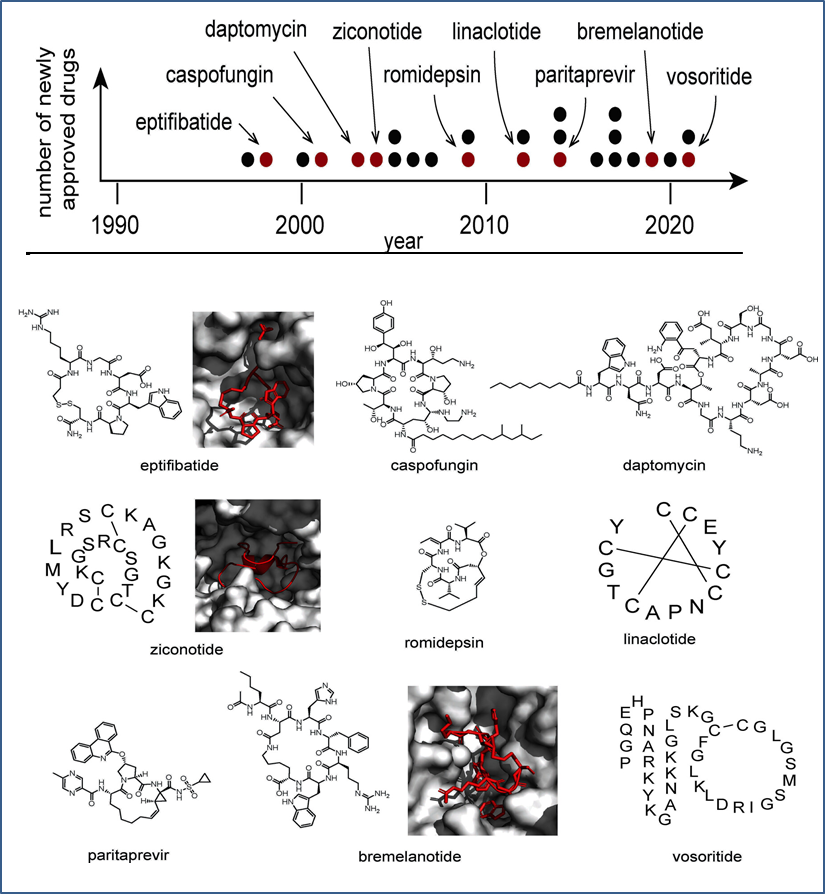
Figure 2: Recently approved cyclic peptide drugs and representative structures.
The clinical success of oxytocin and vasopressin provided early validation of cyclic peptides as drugs. These disulfide-bridged nonapeptides adopt well-defined conformations essential for receptor recognition and signaling. Importantly, cyclization not only stabilized their bioactive structure but also improved their in vivo lifetime relative to linear analogues.
From a discovery chemistry perspective, these molecules demonstrated that even minimal macrocyclization can have profound effects on biological performance, guiding subsequent generations of cyclic peptide design.
Modern discovery platforms have significantly expanded access to cyclic peptide chemical space. Techniques such as combinatorial synthesis, split-and-pool libraries, and display technologies allow rapid generation and screening of structurally diverse macrocycles.
High-throughput screening combined with structure-based design enables efficient SAR development, where changes in ring size, cyclization position, or residue chemistry can be directly correlated with biological outcomes. This iterative synthesis–test–learn cycle is central to cyclic peptide lead optimization.
There is no universal cyclization strategy, each approach influences conformation and physicochemical properties differently:
Traditional methods such as lactam and disulfide bridge formation remain foundational due to their reliability. However, modern approaches—such as click chemistry, thioether formation, carbon–carbon stapling, and metal-catalyzed couplings—enable access to non-natural topologies that can improve permeability, stability, or target specificity. These diverse motifs are summarized in Figure 3, reflecting the expanding toolbox available to discovery chemists.
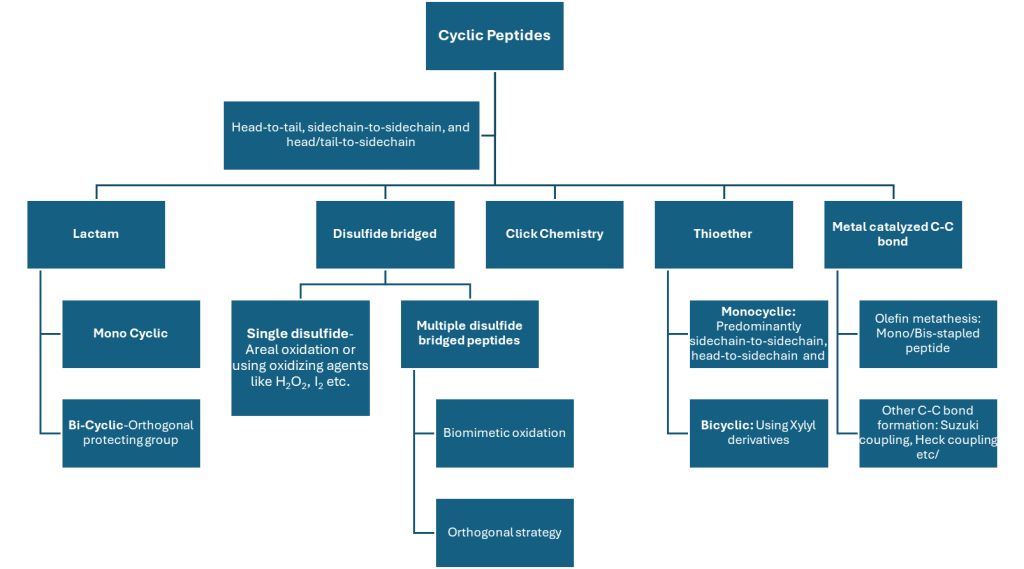
Figure 3: Common cyclization motifs and chemistries used at Aragen.
Chemical modification is often essential to transform potent cyclic peptides into viable leads. Incorporation of unnatural amino acids can enhance binding interactions or reduce metabolic liability. Backbone N-methylation can modulate hydrogen bonding patterns, influencing both conformation and membrane permeability.
Conjugation strategies—such as attachment of lipids, PEG chains, chelators, carbohydrates, or fluorescent probes—extend functionality without disrupting core activity. These modifications can improve exposure, enable imaging, or facilitate targeted delivery, adding significant value during lead optimization.
Early‑stage design choices in discovery chemistry play a decisive role in determining how smoothly a cyclic peptide can progress into development. Factors such as macrocyclization yield, susceptibility to epimerization, choice of orthogonal protecting groups, and purification complexity must be anticipated from the outset to prevent costly bottlenecks later in the pipeline.
As illustrated in Figure 4, translating milligram‑scale exploratory synthesis into multigram or larger quantities requires chemistry that is inherently robust, reproducible, and amenable to scale. When discovery and process chemistry are aligned early, promising cyclic peptides retain their manufacturability as they advance, ensuring that strong biological candidates are not derailed by avoidable synthetic barriers.
Figure 4: Aragen’s capability from milligram to multigram cyclic peptide synthesis.
Aragen’s peptide platform-PeptARx, combines deep expertise in organic synthesis with advanced peptide chemistry capabilities. Key strengths include:
This integrated approach allows discovery teams to push chemical boundaries while maintaining a clear path toward development and commercialization.
Cyclic peptides offer discovery chemists a powerful modality to address complex biological targets with precision and control. Through strategic cyclization, chemical modification, and early consideration of developability, cyclic peptides can be optimized to deliver high potency, stability, and translational potential.
As synthetic technologies and screening methodologies continue to advance, cyclic peptides are poised to play an increasingly central role in drug discovery—transforming challenging molecular concepts into clinically meaningful therapies.