
Surface plasmon resonance (SPR) is a highly versatile technology used to measure molecular interactions in real-time. It works by detecting changes in the refractive index when two molecules bind to one another, allowing detailed insights into binding kinetics without the need for labels or additional chemical modifications. This optical biosensor technology has become indispensable in understanding biomolecular interactions and is widely adopted across the pharmaceutical industry, particularly for applications involving small and large molecule drug discovery.
SPR plays a crucial role throughout the drug discovery and development process, from early-stage screening to lead optimization and mechanistic studies. By providing a label-free, real-time analysis of biomolecular interactions, SPR enables researchers to evaluate complex binding events with unparalleled sensitivity and precision.
At Aragen, our Assay Development and Screening (ADS) team leverages the SPR technology (Biacore™ 8K+) to advance your small and large molecule drug discovery journey. The platform complements our comprehensive portfolio of biochemical and cellular assays, enabling real-time, label-free analyses of a wide range of biomolecular interactions—including protein-small molecule, protein-nucleic acid, protein-peptide, and antigen-antibody. Whether you are targeting kinases, proteases, GPCRs, or challenging protein-protein interactions (PPIs), our SPR experts help you identify and validate novel hit candidates efficiently. Integration of SPR into your drug discovery program will enable rapid identification of hit candidates and, kinetic information will facilitate the ranking of these potential candidates. The various applications of Biacore™ SPR in small and large molecule drug research has been discussed below.
Fragment-based screening has gained prominence as a highly effective strategy in drug discovery, especially in the identification of chemical scaffolds with therapeutic potential. SPR technology allows for high-throughput screening of large fragment libraries (up to several thousand compounds) with minimal sample requirements. This is particularly advantageous compared to traditional techniques such as X-ray crystallography and nuclear magnetic resonance, which typically demands larger quantities of target protein.
SPR excels in fragment screening due to its ability to detect small, low-affinity interactions that are often missed by other methods. The technique’s high sensitivity allows for the detection of real-time binding events from low molecular weight fragments (120–300 Da), despite their typically weak affinities in the millimolar range. This makes SPR a powerful method for early stage hit identification, providing a reliable starting point for subsequent screening steps.
The screening process typically begins with a “clean screen” to eliminate fragments that bind non-specifically to the chip surface. This critical step ensures that downstream data reflects true target-specific interactions, increasing the reliability of results.
Given the low molecular weight of fragments, a high-density immobilization of the target protein is often required to generate detectable signal. To ensure that the immobilized protein remains active over time, surface activity is monitored—preferably using a tool compound, if available—for 24 to 48 hours.
Fragment-based screening via SPR can be performed in two primary modes: direct binding and competition. In the direct binding mode, each fragment is typically injected at a single concentration (mostly 100 μM), and the resulting sensorgrams are analyzed to identify potential binders. SPR analysis software enables in automatic identification of binders and tagging of fragments with atypical binding behaviour for further review.
Since fragment binding often includes both specific and non-specific components, competition screening is used to clarify binding specificity. This involves measuring the binding response of the fragment alone, followed by measurement in the presence of a specific binder. The differential response helps isolate true binding events (figure 1). Parallel inclusion of negative controls during this mode is recommended to further distinguish non-specific binders.
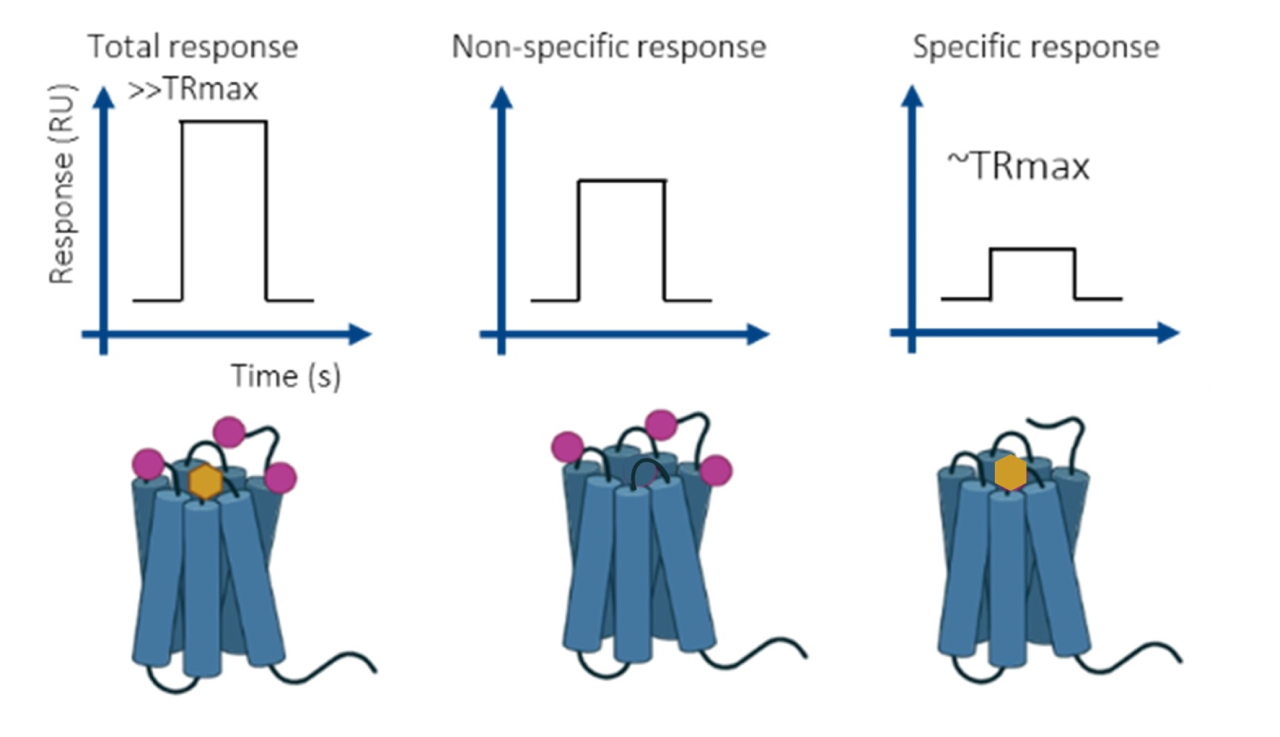
Figure 1: Binding responses in three different run conditions-resolving the non-specific binding issue.
Because the binding signals from fragments are inherently small, it is advisable to include a related but non-target control protein during the screen. This helps in identifying and excluding promiscuous or non-specific binders early in the process.
SPR evaluation software (Bia evaluation software) also enables high-throughput data processing, allowing for efficient filtering of hits and prioritization based on specificity and binding characteristics. Once potential hits are identified, they can be subjected to dose-response analyses, as well as specificity and robustness evaluations.
During the lead optimization stage, SPR is routinely used to determine the kinetic parameters of a small set of compounds to guide structure-activity relationship (SAR) studies. By measuring the key parameters such as the association rate (ka), dissociation rate (kd), affinity (KD) (via equilibrium or kinetic analysis), and the stoichiometry of the interaction, SPR enables researchers to determine the best candidate for further development. Additionally, SPR can measure the residence time (t1/2) of small molecules bound to their targets, a crucial factor in determining drug potency and efficacy. Drugs with slow dissociation rates have been shown to exhibit enhanced clinical efficacy, making SPR a vital tool for optimizing lead candidates for better selectivity, potency, and efficacy.
As drug discovery progresses, understanding the mechanism of action (MoA) of lead candidates becomes critical. SPR technology is widely used in competition assays to differentiate between allosteric and orthostatic binding modes, providing deep insights into the therapeutic potential of drug candidates. Furthermore, SPR plays a significant role in protein-protein interaction inhibitor screening, where it helps identify the compounds that can disrupt crucial signalling pathways, offering valuable information for the design of targeted therapies.
Protein and antibody engineering often involves optimizing the structure and function of therapeutic proteins. SPR technology is widely employed to assess the binding activity and structural integrity of proteins or antibodies after each round of mutation or modification. With its high throughput and sensitivity, SPR allows for rapid screening of large numbers of engineered variants, enabling the selection of the most promising candidates for further development.
Therapeutic monoclonal antibodies (mAbs) dominate the protein therapeutics market, and since the first Biacore™ system launch in 1990, SPR has become central to antibody development workflows. Notably, the bestselling antibody Humira (initially D2E7) was shortlisted from a Biacore™ screen in the mid-1990s.
Throughout antibody discovery and development, SPR provides real-time insights into binding kinetics, specificity, and potency. In early discovery, it is used to screen phage display libraries, identify optimal binding clones, and select candidates with favorable kinetic profiles such as low dissociation rates (Koff). SPR also supports epitope binning, grouping antibodies by binding sites to streamline candidate selection and reduce development costs.
Beyond interaction analysis, SPR is valuable for assessing developability parameters including aggregation risk, solubility, immunogenicity, and solution stability. This ensures that antibody leads are not only potent but also manufacturable and clinically stable, making SPR indispensable across the entire therapeutic antibody pipeline.
Surface Plasmon Resonance technology has become an essential tool in the drug discovery and development landscape. Its ability to provide real-time, label-free monitoring of molecular interactions with high sensitivity makes it invaluable for applications ranging from fragment screening to lead optimization and antibody engineering. As the Contract Research, Development, and Manufacturing Organization sector continues to advance, SPR technology will play a pivotal role in accelerating drug development by delivering critical insights into binding kinetics, specificity, and mechanism of action.
At Aragen, we offer over a decade of expertise in SPR (Biacore™ 8K+) to accelerate your drug discovery programs. Our skilled scientists are proficient in executing large library screenings, delivering fast, reliable insights into compound binding kinetics. We offer:
By partnering with Aragen, you gain access to cutting-edge SPR capabilities designed to meet the evolving demands of the pharmaceutical industry—helping you make confident, data-driven decisions at every stage of discovery.