
In today’s development landscape, the distance between a promising molecule and a first-in-human trial is no longer defined by time alone — it is defined by integration. The transition from discovery to an Investigational New Drug (IND) submission is not a procedural milestone; it is a scientific stress test.
For emerging biotech companies and global pharma alike, IND-enabling pharmacology represents a pivotal inflection point. It is here that early biological promise must be translated into regulatory-grade confidence. The question is no longer whether a molecule works in a model — but whether it can withstand the scrutiny of exposure alignment, safety margins, translational biomarkers, and manufacturing reproducibility.
At a strategic level, IND-enabling pharmacology is the deliberate engineering of a bridge. That bridge rests on four structural pillars: DMPK, bioanalysis, toxicology & safety pharmacology, and pharmaceutical development (Figure 1). But pillars alone do not create connectivity. Translation happens in the integration between them.
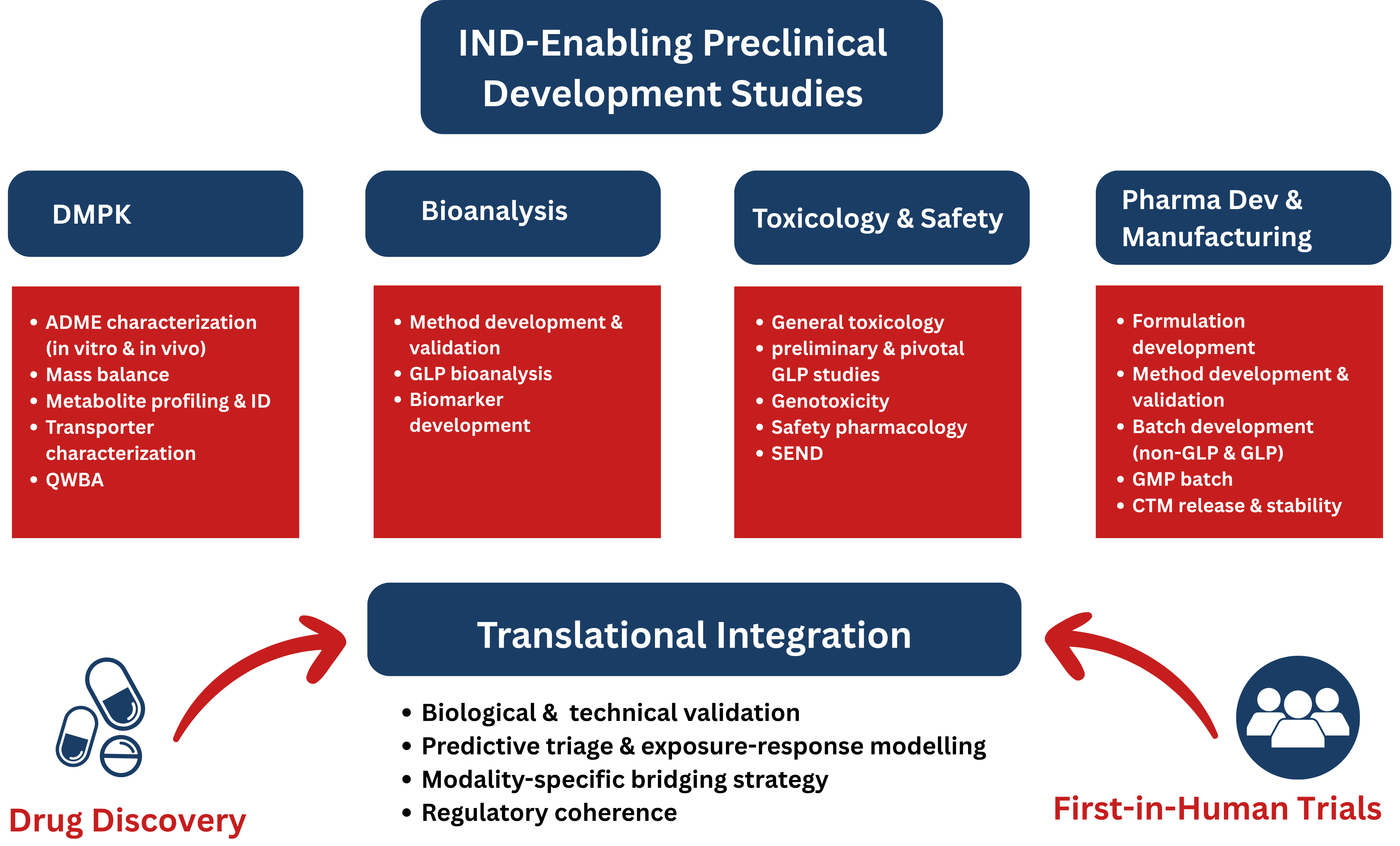
Figure 1: The integrated framework of IND-enabling pharmacology bridging drug discovery to First-In-Human (FIH) trials.
Many programs approach IND-enabling studies as a checklist exercise. In practice, success depends less on completing studies and more on aligning them.
A development-ready program demonstrates:
Without cross-functional coherence, even high-quality datasets can fail to support a defensible clinical hypothesis.
A CRDMO’s value at this stage is not simply operational capacity — it is architectural oversight. Integrated program design prevents late-stage misalignment that can delay or derail IND submission.
Strong IND-enabling programs begin well before formal safety studies. Biological validation must be accompanied by translational foresight.
It is not enough to confirm that target modulation alters disease biology. The program must anticipate:
Early integration of DMPK, bioanalytical strategy, and safety considerations reduces downstream surprises. For complex modalities — including biologics, gene therapies, and RNA therapeutics—species limitations and target-mediated drug disposition require even earlier cross-disciplinary planning.
Modern drug discovery depends on structured triage — a staged test cascade of increasing biological complexity. However, the most effective triage is predictive rather than eliminative.
Compounds advancing toward IND readiness must demonstrate more than potency. They must show:
At the IND-enabling stage, refinement is as important as elimination. Medicinal chemistry optimization guided by PK/PD modeling ensures that candidates entering toxicology are not only biologically promising but developmentally viable.
The CRDMO advantage lies in synchronizing these decision gates across disciplines rather than operating them in isolation.
The most critical integration point in IND-enabling pharmacology is exposure–response alignment. Regulators expect to see a clear, quantitative relationship between:
Establishing this alignment requires tight coordination between DMPK scientists, pharmacologists, bioanalytical teams, and toxicologists. Exposure levels driving efficacy must be directly comparable to those defining the No Observed Adverse Effect Level (NOAEL).
PK/PD modeling then becomes the predictive engine that connects animal data to human starting dose justification. Without this quantitative backbone, translation becomes assumption-driven rather than evidence-based.
The bridging approach to IND-enabling studies shifts significantly depending on the nature of the therapeutic (Table 1). Small molecules (New Chemical Entities) generally follow a standardized, well-trodden path, whereas Biologics (New Biological Entities, such as monoclonal antibodies) require a more “case-by-case” strategy.
For small molecules, translation challenges often center on metabolic variability, CYP interactions, and standardized safety pharmacology requirements. Two-species toxicology and genotoxicity batteries are well defined, but exposure prediction and drug–drug interaction risk must be carefully modeled.
Biologics, by contrast, present distinct complexities: limited relevant species, target-mediated drug disposition, and immunogenicity considerations such as anti-drug antibodies. Safety pharmacology is frequently integrated into toxicology studies, and exposure interpretation must account for nonlinear kinetics.
| Feature | Small Molecules (NCE) | Biologics (NBE/Mabs) |
| Species Selection | Two species (one rodent, one non-rodent). | Only pharmacologically relevant species (often NHP). |
| Metabolism Focus | CYP450 enzyme inhibition and induction studies. | Degradation into peptides and amino acids; Focus on “TMDD”. |
| Safety Pharmacology | Standardized “Core Battery” (CNS, CV, Respiratory). | Often integrated into general toxicology studies. |
| Immunogenicity | Rarely a primary concern in preclinical stages. | Critical; assessment of Anti-Drug Antibodies (ADA). |
| Genotoxicity | Standard battery (Ames test, Chromosomal aberration). | Generally, not required unless there is a specific concern. |
An effective IND-enabling strategy adapts to these modality-specific realities rather than applying template solutions. Integrated CRDMO teams that understand both chemical and biological modalities reduce regulatory uncertainty and compress timelines.
The increasing adoption of New Approach Methodologies (NAMs) reflects both scientific evolution and regulatory openness to innovation. Advanced in vitro systems, organ-on-chip platforms, computational modeling, and in silico PK prediction tools are strengthening translational accuracy.
However, their greatest value lies in integration. NAMs enhance early triage, refine mechanistic insight, and support exposure modeling — but must be aligned with traditional in vivo data to support regulatory acceptance.
For highly human-specific modalities, NAMs and humanized systems are becoming essential. Their thoughtful incorporation can de-risk programs before formal toxicology investment begins.
An IND submission is evaluated not as a collection of studies but as a coherent scientific narrative. Regulators expect logical progression from target identification through exposure modeling to dose justification.
Key elements of a defensible package include:
Early regulatory engagement through pre-IND interactions further reduces uncertainty and strengthens submission strategy. By integrating PK/PD modeling early in the process and engaging in Pre-IND meetings with regulatory bodies, drug developers can mitigate the risk of a “Clinical Hold” and ensure that the bridge to human trials is built on a foundation of rigorous, reproducible science. A CRDMO operating with regulatory foresight helps sponsors anticipate questions rather than react to them.
IND-enabling pharmacology studies are designed to reduce uncertainty before first-in-human exposure. Through disciplined triage, model progression, PK/PD integration, and increasingly the use of NAMs, developers construct a scientifically defensible bridge from target validation to clinical dosing.
While the core objective remains consistent—demonstrating safe and rational readiness for human trials—the pathway differs notably between small molecules and biologics. Submission of an IND to the U.S. Food and Drug Administration is therefore not merely a regulatory step, but the culmination of an integrated pharmacological strategy that aligns discovery science with clinical translation.
Turn complex science into confident decisions. Partner with Aragen BioSafety team today.